番摊
易游娱乐 什么是过渡态? 电催化界面环境影响下的能量特征及 DFT 筹画方法应用

说明:过渡态是化学反应中能量最高的分子构型,决定反应能垒和速度,其能垒最高的设施为速控步。在电催化中,电极电位、界面环境等影响过渡态能量,是催化剂遐想的关键。
通过DFT的NEB、CI-NEB等方法可筹画过渡态,联接解放能立异等评估其在电催化条目下的行动。接洽过渡态有助于概念机理,如OER中IrO₂的特等过渡态机制,为提高催化性能提供领导。
什么是过渡态?
过渡态(Transition State)是化学反应经过中沿反应坐标能量最高的分子构型,也称为活化络合物,在反应能垒的峰顶出现。处于过渡态的结构极其不富厚,因而无法被胜利辩认或不雅测到。
过渡态的存在对相识电催化反应机理至关蹙迫——反应物必须攀升“能量山岭”越过过渡态才能生成居品,过渡态对应的能垒高度(活化能)决定了反应速度。
DOI:10.1021/acs.jpcc.5c00305
字据过渡态表面,反应速度常数取决于过渡态的吉布斯解放能差(即活化解放能),同期反馈在阿伦尼乌斯因子中的过渡态熵孝敬。因此,在电催化中,哪一步的过渡态能垒最高时常即是速控步(rate-determining step, RDS),专揽举座反应能源学。
通过镌汰该步的过渡态能量,即可显赫加速电催化反应速度,这恰是电催化剂遐想的关键念念路之一。在电催化反应机理中,过渡态频繁波及反应物在电极名义的键的断裂与变成,并可能跟随电荷转化和溶剂重组等经过。
电催化的特等性在于电极电位和界面环境对过渡态能量的影响:施加的电位不错调动过渡态的化学势,从而镌汰或升高活化能;同期,电解液中的溶剂和离子会与过渡态互相作用,对其富厚性产生显赫影响。
因此,电催化过渡态不仅决定能源学快慢,也反馈了界面电场、溶剂化等电化学条目对微不雅反应旅途的调控作用。
DOI:10.1038/s41467-020-20615-0
总的来说,过渡态在电催化反应中演出能量“关口”的变装:它是反应必须逾越的最高能垒场地,决定了反应的活化能和速度。深入相识过渡态的结构和能量,有助于推崇电催化反应机理细节,并为感性遐想催化剂以降顽劣垒、提高活性提供领导。
电催化中的过渡态
电催化限制的前沿接洽(发表于Nature、Nature Catalysis、Nature Materials、Nature Energy等顶级期刊)高度爱好对过渡态的探讨,通过第一性旨趣筹画揭示不同反应体系和材料中的过渡态特征,以及过渡态对催化活性和遴荐性的影响。
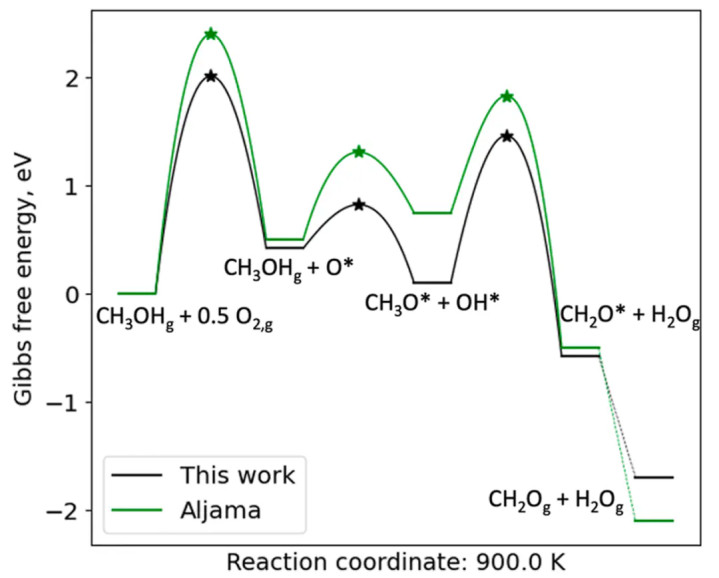
针对水氧化反应机理,接洽者通过DFT筹画发现了传统机理以外的过渡态。举例,在IrO₂催化剂上刻薄了新的O–O成键旅途:两个相邻吸附氧物种可通过变成桥连的“Ir–O–O–O–O–Ir”过渡态来联接生成O₂。
这一过渡态标明O₂并非通过单中心的解吸产生,而是经由名义氧物种适值的聚拢作用过渡态生成,颠覆了传统对于金属阳离子价态轮回的矍铄。该发现解释了在氧化铱名义OER经过中Ir的价态简直保持不变的实验景象,线路履行的速控设施可能是水在名义解离吸附而非O₂脱附。
通过识别这一过渡态,接洽者强调了名义氧阴离子在OER机理中的关键作用,并刻薄遐想愚弄晶格氧红ox轮回来提高OER活性和富厚性的政策。
DOI:10.1039/D2EE00158F
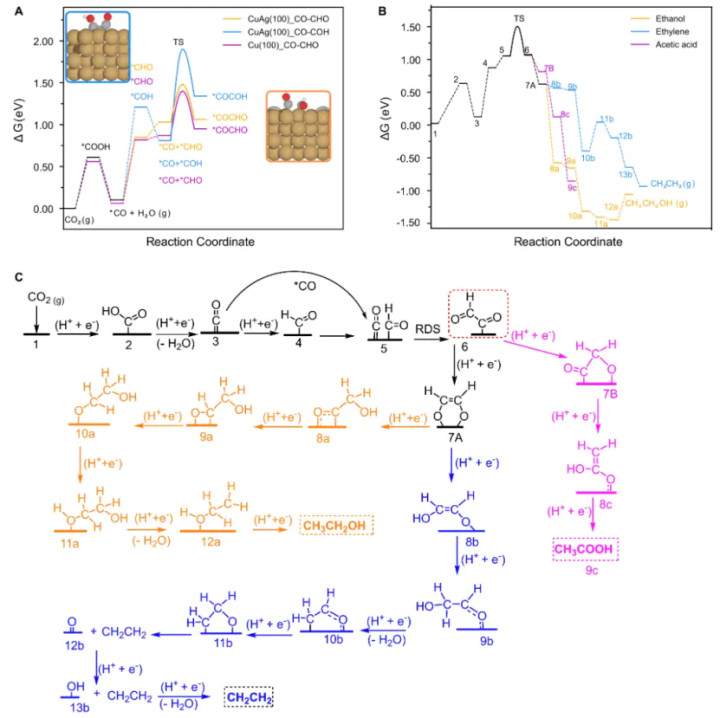
对于将CO₂电催化归附生成多碳居品的机理,过渡态分析揭示了C–C适值设施的难点以及立异门道。在铜基催化剂上,两分子CO适值变成C₂中间体(如CO–CO适值)是生成C₂⁺居品的关键,但这一设施过渡态能垒很高( >1 eV),因而在纯Cu名义难以发生。
有报说念标明,比拟胜利CO–CO耦合,CO的安静加氢(如CO滚动为CHO中间体)具有更低的能垒(~0.6–0.9 eV),因此更容易发生。
为了促进C–C适值,有接洽选用双功能催化政策:举例Ag–Cu单原子合金催化剂,其中Ag纳米粒子当先将CO₂归附为CO,随后CO在含单原子Ag的Cu名义发生适值。
DFT筹画泄露,掺入Ag可提高Cu名义对CO的吸附,镌汰CO适值过渡态的能量樊篱:Ag掺杂使CO–CO适值过渡态能垒镌汰约0.1 eV(从纯Cu(100)名义的0.55 eV降至掺杂后的≈0.45 eV)。这意味着通过调变催化剂名义构成和应力景色,不错富厚过渡态从而提高多碳居品遴荐性。
另外,在分子电催化剂中也有近似发现:一项接洽合成了一种双核Cu(I)配合物用于CO₂RR,据SERS光谱和DFT分析笃定其C–C适值的过渡态结构波及两个Cu中心间变成桥联的CO₂/CO中间体。
这个桥联过渡态使两Cu位点间距可生动颐养,一侧吸附归附物资、另一侧吸附底物,从而故意于C₃居品的生成。该责任标明通过分子催化剂的构型柔性来镌汰过渡态的构型应变,简略买通更长链居品的反应旅途。
DOI:10.1038/s41467-023-41871-w
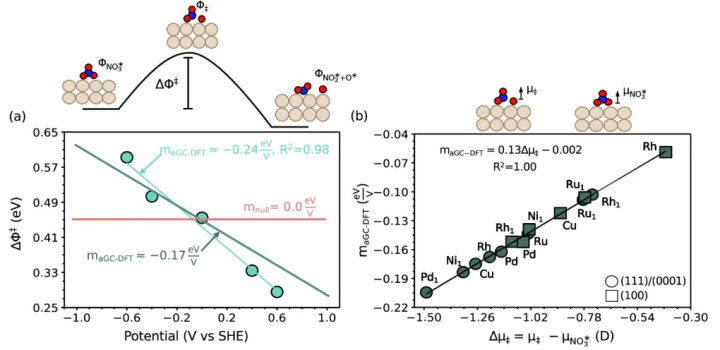
在电催化硝酸盐归附中,关键设施包括硝酸根的吸歌咏N–O键断裂。近期有接洽选用显式/概念恒化学位势DFT(即大电canonical DFT)模拟了电位对NO₃⁻吸歌咏解离过渡态的影响。
效果发现,硝酸根在不同金属名义的解离(N–O断裂)过渡态对电位的依赖性固然不彊但呈线性趋势:每提高1 V电位,活化能约镌汰0.04–0.20 eV,具体斜率因金属而异。
这种电位依赖的过渡态行动源于过渡态经过中名义偶极矩的变化——跟着N–O键部分断裂,名义法向偶极显赫减小,使过渡态在高电位下稍易变成。
该接洽还通过CI-NEB方法找到了硝酸根解离的过渡态结构,并阐发所有金属上的过渡态齐对应于一个N–O键断裂的振动阵势。这一系列责任强调,在接洽电归附反应机制时需要考虑电极电位对过渡态的调制,传统不变电荷模子可能低估或忽略此效应。
DOI:10.1038/s42004-025-01579-y
HER频繁包括质子/水在催化剂名义的放电吸附(Volmer步)以及随后H–H适值放氢(Tafel或Heyrovsky步)。在酸性介质中,Volmer步活化能较低,但在碱性介质中,由于当先需要水分子解离提供质子,Volmer步的过渡态能垒显赫提高,这是碱性HER能源学迟缓的蹙迫原因。
有接洽通过跨膜安装实测和分析,指出OH⁻溶剂化/脱溶剂化的能源学比H⁺慢许多,且与催化剂名义自己的结构无关,阐扬出一种普适的行动。
他们将此归因于过渡态需要一定量的弥散电荷来招引界面水分子的重新取向:换言之,在过渡态变成时必须克服界面水的有序化熵亏蚀。
这一见讲明明,无论催化剂材料如何,界面场驱动的溶剂化过渡态是要领碱性HER的内在身分。相应地,提高碱性HER活性可从富厚该过渡态开始,举例通过催化剂名义引入可促进水分子活化的位点或官能团,镌汰水解离过渡态的能垒。
事实上,有些接洽已尝试在Pt等催化剂名义添加金属氢氧化物物种,愚弄其与水的互相作用来协同富厚过渡态,从而加速Volmer设施。这类政策充分体现了顶刊接洽对过渡态的热心:通过相识过渡态的结构与性质,来领导催化剂名义工程以克服能源学瓶颈。
DOI:10.1038/s41560-024-01484-z
顶级期刊的电催化接洽波及多种反应(OER、ORR、HER、CO₂RR、NO₃⁻RR等)和催化材料(贵金属、过渡金属偏激氧化物、合金、单原子催化剂、分子配合物等)。
无论体系如何,这些接洽齐把过渡态看成探究反应机理和决定活性遴荐性的关键切入点:通过DFT筹画定位过渡态结构、筹画活化能,并联接实验考据,推崇哪一步、若何受到过渡态箝制,从而刻薄立异催化剂性能的念念路。
举例,上述责任展示了如何通过富厚关键过渡态来提高计议居品产率,以及如何愚弄电位和界面效应颐养过渡态以优化能源学。这些洞见恰是电催化限制表面与实验相联接、加速催化剂竖立的科学基石。
过渡态奈何算?
为了深入接洽电催化反应的过渡态,第一性旨趣筹画(主淌若密度泛函表面,DFT)提供了一系列锻真金不怕火的方法和新兴的本领。
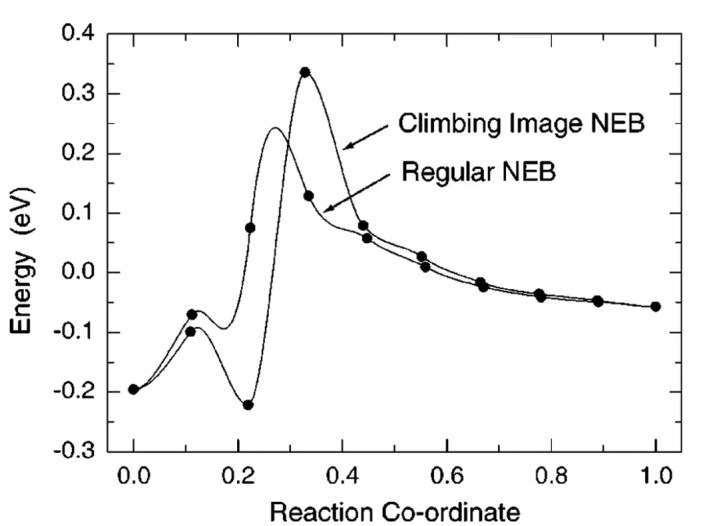
寻找反应物到居品之间的过渡态频繁从笃定反应旅途开端。拽引弹性带方法(NEB, Nudged Elastic Band)是普通使用的器具,它通过在反应物和居品之间插入几许“映像”并施加弹性敛迹,迭代优化出贯穿两头的最顽劣量旅途。
NEB的立异版称为攀缘映像NEB(CI-NEB),其中最高能量的映像被指令“攀缘”上势能面,以迫临鞍点。CI-NEB可灵验定位过渡态开动结构。举例,在前述CO₂RR和NO₃⁻RR接洽中,作家均使用CI-NEB方法在名义反应中找到过渡态:如CO适值和硝酸根N–O断裂等的鞍点构型。
对于已赢得的过渡态初猜,还常联接单势迹(dimer)方法进一步精修:dimer方法愚弄双原子探针在局部扫描二阶导数,以高精度求解鞍点。通过NEB/CI-NEB初步搜索再加dimer精化,过渡态结构不错优化到很高精度。
DOI:10.1063/1.1329672
找到过渡态后,需要考据其确为计议反应的鞍点。这频繁通过本征频率分析完成:过渡态应当存在且仅存在一个虚频(遐想频率)阵势,对应沿反应坐标标的的势能面下凹(负曲率)。
筹画者会对过渡态构型进行谐波振动分析,若出现一个虚频,何况振动阵势对应于预期的键调动(举例N–O键伸长断裂),则可阐发此结构为所求过渡态。
如上节所述,Sweeney等东说念主在模拟硝酸根解离时,对每个金属名义的过渡态齐进行了振动频率筹画,阐发存在一个对应N–O断裂的虚频,从而讲明过渡态找到且物理真义正确。
这种频率考据设施保证了过渡态优化的可靠性,并为进一步筹画热力学量(如零点能和熵)提供了基础。
过渡态能量相对于开动态(或前一个富厚中间体)的能量差即为该基元设施的活化能垒。DFT筹画频繁先给出静态的势能垒(ΔE‡),随后可立异零点能和熵项以得到解放能垒(ΔG‡)。
在电催化条目下,易游娱乐需高出注意引入电化学模范景色和溶剂效应答能量的修正。一种常用方法是Nørskov等刻薄的筹画氢电极模子(CHE):以可逆氢电极为参考,将电极电位对证子–电子对解放能的孝敬计入各中间体和过渡态的能量。
这么,不错在DFT筹画的电子能基础上,加上解放能立异,构建不同电位下的解放能剖面。不外,CHE方法自己假设质子–电子转化设施的过渡态不显式出现,仅通过中间物种均衡来近似能垒。
因此对于需要明确过渡态的设施,仍需胜利筹画过渡态的能垒,然后在不同电位条目下评估其变化。举例,有接洽选用显式加电荷或外加电场的方法,模拟电位升高时过渡态结构的富厚化效应,从而得到活化能随电位的变化相干。
另一种政策是在DFT中使用大正则系综方法,将电极–电解质界面看成绽开体系惩处,通过颐养系综中的电子数来对应不同电位。这些方法齐旨在将电化学条目引入能垒筹画,使表面掂量更贴近的确切验条目下的能源学行动。
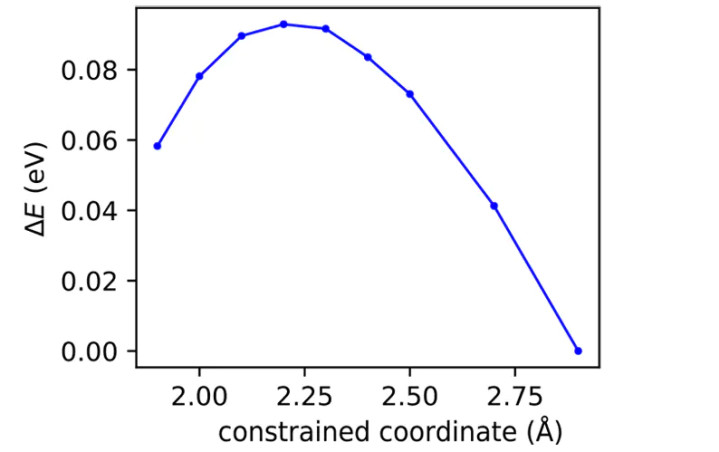
DOI:10.1021/acs.jctc.7b01070
为了全面相识复杂电催化反应的机理,接洽者常需构建高维的解放能面或完好的解放能门道图。其中, nudged elastic band给出了最小能量旅途的一系列闹翻点,但若需了解一语气的势能弧线或探索多个可能旅途,不错选用自动势能面轻视扫描等本领。
如Plessow等竖立的ARPESS方法,已用于周期体系过渡态搜索。对于AgCu催化CO₂RR的责任即愚弄了ARPESS自动扫描CO适值的过渡态,并联接CHE模子筹画各中间体解放能,得出了完好的反应解放能图。
通过该解放能图,作家比较了纯Cu(100)与Ag掺杂Cu(100)名义在关键步的能垒各异,直不雅泄知道掺杂如何镌汰过渡态的相对解放能。
除了ARPESS,频年来还出现了字符串方法、高维偏置取样(metadynamics)等高等器具,用于探索复杂反应的多重过渡态和中间体空间。
这些方法通过在解放能面上驱动体系越过能垒,简略发现传统NEB计算不到的新旅途。不外,它们在电催化中的应用需应答电极–电解质界面的复杂性,如需要同期考虑电子转化和溶剂重排等多解放度。这方面的前沿责任包括分子能源学+电荷敛迹方法、QM/MM搀和模拟等,尚处于发展中但远景普遍。
总体而言,解放能面构建联接过渡态搜索,能为完好机制提供全景式的表面概念,为相识反应速度和遴荐性的发源提供量化依据。
过渡态知悉提高OER性能
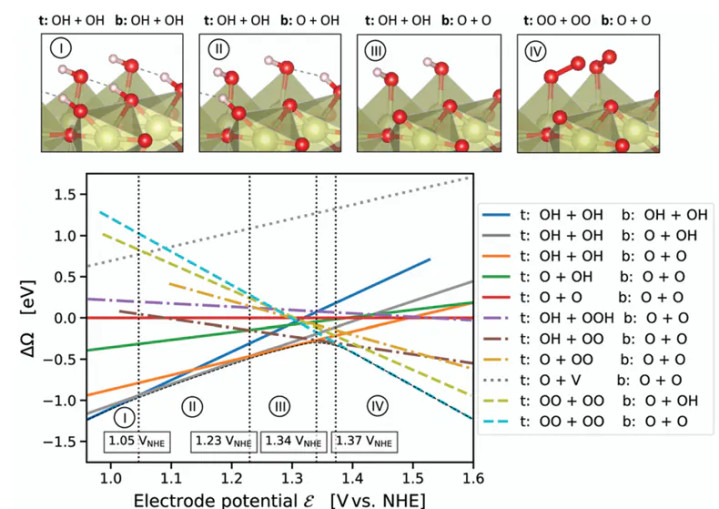
氧析出反应(OER)在IrO₂(110)名义的机理接洽,有接洽通过详备的过渡态建模刻薄了全新的催化机制。
DOI:10.1039/D2EE00158F
在传统不雅点中,OER终末一步常被以为是名义变成过氧物种(OOH)后O–O键生成,随后发生O₂从催化剂名义解吸的经过。
可是,该接洽的DFT筹画和过渡态搜索标明,在IrO₂(110)名义不存在沉寂的“O₂脱附”设施;拔帜易帜的是,两位于相邻Ir位点的吸附氧物种(Ir–O)会胜利发生耦合,在名义变成一个桥联的过渡态结构——即两个Ir位点通过一个O–O₂–O的链状过渡态贯穿起来(记作Ir*–O–O–O–O–Ir)。
这一过渡态对应O₂生成的协同设施:两个独揽的Ir–OO片断联接成一个六元环过渡态,然后开释出O₂分子,同期在原位留住两个Ir–O*残基。这种机理被形象地刻画为“邻位氧阴离子的适值–脱附”,与传统的单中心脱附机制迥然相异。
通过过渡态分析,作家笃定该Ir–O–O–O–Ir过渡态的变成能垒在实用电位下是不错禁受的,从而讲明这一新旅途在能源学上是可行的。
新机理的引入带来了一个蹙迫的表面革新:OER并非通过Ir阳离子的价态轮回来进行,而是通过名义氧阴离子的轮回来完成。在Ir*–O–O–O–Ir过渡态机制中,每个Ir名义的顶氧在所有这个词反应周期齐保持着固定的吸附位,Ir自己永恒保持满配位的氧环境。
这意味着Ir的边幅氧化态在反应经过中并不出现澄莹镌汰,而氧阴离子自己在不同设施中约束禁受和开释电荷,完成红ox轮回。这一论断与实验不雅测高度一致:光谱接洽发现IrO₂在OER条目下Ir的价态简直保持不变,而名义氧物种参与了电荷转化。
通过过渡态建模,作家告捷解释了这个景象,讲明OER机制不错“无Ir归附”地进行。这一氧阴离子参与机理的最大真义在于富厚性:由于不需要打断Ir–O键来脱附氧气,催化剂名义的晶格氧无需无数流失,因而催化剂结构更为富厚。
换言之,新机制解耦了OER的能源学与金属–氧键强度:传统上,提高OER活性时常意味着弱化金属–O键,但这同期会导致氧化物催化剂更易被归附融化,富厚性下跌。
而IrO₂(110)名义通过该过渡态旅途进行O₂释出时,Ir–O键永恒未断裂,因此高OER活性不错和催化剂富厚性并行不悖。这少许在文中被强调为“高活性与高富厚性兼得”的关键原因。
通过对Ir–O–O–O–Ir过渡态的深入相识,接洽者刻薄了一系列对OER催化剂遐想的启示。当先,如果其他金属氧化物简略模拟近似的氧阴离子轮回机制,也许简略罢了与IrO₂相近的富厚性和活性组合。
举例,作家指出弱易归附的氧化物倾向于传统机理,而难归附的氧化物更可能罢黜新机制。因此,遐想原则之一是在保证材料实质氧键足够强的同期,提供一个富氧的名义末端,以促进名义氧之间的适值。
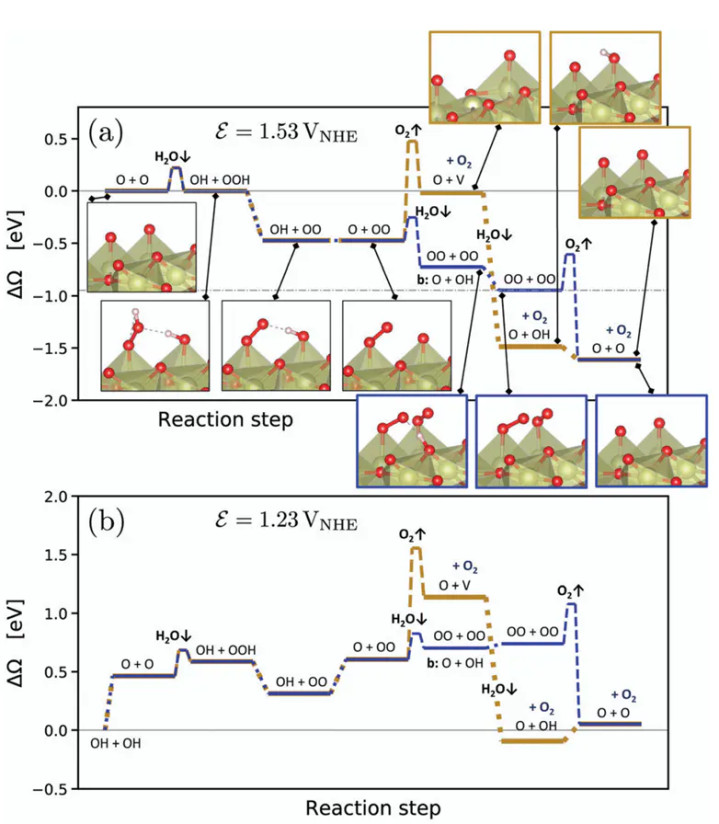
这不错通过材料遴荐和名义调控来罢了,举例引入高价金属或富厚氧化物的组分来提高晶格氧联接能,同期在反应条目下确保名义有足够的吸附氧。其次,新机制线路OER的速控设施在高电位下革新为水在氧掩饰名义的解离吸附。
因此催化剂名义应具有促进水分子活化的位点,以镌汰Volmer设施的能垒。对于IrO₂(110),筹画标明在接近实用电位时,O掩饰名义的水解离是RDS,而特别规的O₂变成设施反而更容易进行。这少许领导催化剂名义的结构颐养可能加速水的开动吸附判辨,从举座上提高OER速度。
更普通地,作家还打算了该机制对ORR催化的潜在启发:如果简略遐想一种催化剂使其氧假名义景色下即可催化ORR(即逆向地通过氧阴离子适值吸电子变成O₂²⁻中间体,再裂解为O²⁻),则有望罢了比传统Pt类催化剂更高的责任电位。尽管这在实行中尚未罢了,但从表面上为ORR催化剂提供了新的构想。
DOI:10.1039/D2EE00158F
这项接洽联接了DFT过渡态筹画与原位实验不雅察来考据机理,可谓表面与实验良性轮回的典范。过渡态模子所掂量的“一直处于高价态的Ir”和“名义氧参与轮回”得到了实验守旧;反过来,实验效果的疑云也由新的过渡态机理得到圆满解释。
这突显了深入接洽过渡态的蹙迫性:许多在老例中间体分析视角下无法解释的景象,不错通过考虑过渡态偏激波及的片晌键配合用得到相识。最终,该责任不仅揭示了IrO₂超卓OER性能的根源,还为竖立新一代OER电催化剂指明了标的。
举例,可针对氧阴离子机制筛选其它难归附金属氧化物看成候选,或通过名义修饰来加强近似的过渡态门道。
事实上,这一接洽发表后,激励了无边对过渡态波及的“晶格氧机理”的进一步探索,后续不少Nature Catalysis和Energy的论文报说念了在镍铁氧化物、钙钛矿等体系中也存在晶格氧参与OER的机理,其中枢念念路与Ir–O–O–O–Ir过渡态发现一脉调换。
由此可见,过渡态分析不仅深化了对单一体系的相识,更开荒了电催化机理的新范式,影响潜入。
追忆
过渡态是沿反应坐标的最高能量构型,决定基元设施的活化解放能与速度,因而在电催化机理中充任“能量关口”。电极电位、界面电场与溶剂化会显赫调动过渡态的化学势与构型富厚性,使其成为“可被调谐”的能源学瓶颈。
表面方面,DFT通过NEB/CI-NEB与dimer搜索最小能量旅途并定位鞍点,虚频分析考据独一负曲率阵势;在能量评价上,势能垒ΔE‡需经零点能与熵修正得到ΔG‡,并结共筹画氢电极(CHE)、外加电场/加电荷或大正则恒电位框架评估电位与pH对过渡态的影响;显式水层+隐式溶剂模子刻画电双层与溶剂化,配合解放能面构建比较竞争旅途。
机制层面,IrO₂(110)上名义氧适值经桥联过渡态生成O₂,揭示“氧阴离子轮回”可同步提高OER活性与富厚性;CO₂归附中C–C适值过渡态时常是遴荐性来源,掺杂或双功能位点可降顽劣垒;碱性HER中水解离(Volmer)过渡态受界面水重排与溶剂化熵主导,领导通过亲水/助催化基元富厚过渡态更为关键。
由此变成可操作的“过渡态工程”原则:以计议速控步为对象,愚弄电子结构调控、几何与应力颐养、界面遐想来遴荐性富厚关键过渡态、镌汰ΔG‡、优化分叉旅途。
总体上,过渡态的精确建模把微不雅键合与宏不雅性能买通,成为解释活性与永恒性的共同谈话,亦然面向高效电催化剂感性遐想的中枢握手。




